|
Help Topics
A. Overview - general information on DOMINE
B.
Getting started - how to use the database, searching DOMINE
C. Terms of
Use
D. Data Download
E. Citing DOMINE
F. Frequently Asked Questions (FAQ)
A. Overview
DOMINE is a database of known and predicted protein domain
(domain-domain) interactions. It contains
interactions
inferred from PDB entries, and those
that are predicted by
13 different computational approaches using
Pfam domain definitions. The domain-domain interaction (DDI) data in the
DOMINE database was gathered from 15 different sources altogether.
|
Data source or method |
Number of interactions |
Comments |
|
iPfam |
4,030 |
iPfam is a database of DDIs that are observed in
PDB entries. Interaction data, dated 17 Feb 2007, was used. |
|
3did |
6,066 |
3did is a collection of
DDIs in proteins for which high-resolution
three-dimensional structures are known. Interaction data, downloaded in Sep 2010
was used. |
|
ME |
2,391 |
ME refers to Lee et al.'s
integrated approach (2006) for the prediction of
DDIs. This method used a Bayesian approach to integrate domain
interactions predicted using a maximum likelihood estimation (MLE)
approach on yeast, worm, fruitfly and human protein interaction networks
with the gene ontology and domain fusion data to arrive at a total
of 2,391 high-confidence predictions. |
|
RCDP |
960 |
Jothi et al.'s Relative
Co-evolution of Domain Pairs (RCDP) approach uses sequence co-evolution
to predict the domain pair that is most
likely to mediate a given protein-protein interaction.
Given a protein-protein interaction, RCDP computes the degree of
sequence co-evolution between all pairs of domains between the two
proteins, and predicts the domain pair with the highest degree of
co-evolution to be the mediating domain pair.
The set of 960
DDIs predicted from 1,180 yeast protein-protein
interactions was used. |
|
P-value |
596 |
"P-value" refers to Nye et
al.'s statistical approach that assigns p-values to pairs of domain
superfamilies, measuring the strength of evidence that this pair of superfamilies form contacts within a set of
protein interactions. A set of p-values is calculated for
SCOP superfamily pairs based
on a pooled data set of interactions from yeast. These p-values were
then used to predict which domains come into contact in an interacting
protein pair. This scheme was applied on protein complexes in the
Protein Quaternary Structure (PQS)
database to predict domain-domain contacts for 705 interacting protein
pairs. Since interactions were predicted between SCOP domains,
SGD was used to map SCOP
domains to Pfam domains, for every yeast protein used, and convert 705
interactions between SCOP domain families to 596 DDIs
among Pfam domain families. |
|
Interdom |
2,768 |
Domain-domain
interactions inferred using domain fusion hypothesis as inferred by Ng et al.
Interaction data from version 1.1, released on 15 Feb 2003, was used. |
|
DPEA |
1,812 |
Riley et al.'s Domain Pair Exclusion
Analysis (DPEA) is a statistical approach to infer DDIs
from the incomplete sets of protein-protein interactions
from multiple organisms. It employs an expectation maximization
algorithm to obtain a maximum likelihood estimate or the probability of
interaction of each potentially interacting domain pair. For each
potential domain pair, a change in likelihood, expressed as a log odds
score, is computed by excluding this domain pair from being considered
as a potentially interacting domain pair. The set of 3,005 domain pairs with log odds
score ≥ 3.0 were designated as high-confidence interactions.
Since only interactions between Pfam-A domains are being considered,
1,193 interactions in which at least one of the interacting partner is a
Pfam-B domain were discarded, reducing the number of interactions to
1,812. |
|
PE |
2,588 |
Guimaraes et al.'s Linear
Programming approach is an optimization approach, which relies on
the parsimonious explanation (PE)
that "DDI
partners are predicted by identifying the minimal weighted
set of domain pairs that can justify a given protein-protein interaction
network". Given a protein-protein interaction network, the PE approach
computes a LP-score, in the range (0,1), for every domain pair that
could possibly justify interaction between two proteins.
False positives in the protein-protein interaction
network are handled using a probabilistic construction (p-scores).
Domain pairs with an LP-score above a certain threshold are considered
as interacting. A set of 3,499 domain pairs with LP-score ≥ 0.5 and 0.0
≤ p-score ≤ 0.1 was used. Since only interactions between Pfam-A domains
are considered, 911 interactions in which at least one of the
interacting partner is a Pfam-B domain were discarded, reducing the
number of interactions to 2,588. |
|
GPE |
1,563 |
GPE builds upon the PE approach
by unifying domains that always occur together in a protein as a singular
'supra -domain', and uses the linear-programming framework as used by PE.
GPE was applied on the redefined Riley et al. dataset (Guimaraes dataset), and the
set of interactions only between Pfam-A domains with LP-score ≥ 0.60 and pw-score
≥ 0.01 was used. Supra-domains were expanded back to individual Pfam-A domains
to obtain 1,563 interactions.
|
|
DIPD |
2,157 |
DIPD constructs feature vectors for
each protein pair within the sets of PPIs (Riley et al. dataset) and non-PPIs, and
uses a discriminative classifier to identify the minimum set of domain pairs/triplets
that can discriminate PPIs and non-PPIs. Each selected feature (domain pair) is
a putative DDI. The sets of predictions on input datasets from Jothi et al.,
Riley et al., and Guimaraes and Przytycka were used to predict a combined 2,157
interactions.
|
|
RDFF |
2,475 |
Chen and Liu's Random Decision Forest
Framework (RDFF) approach explores all possible DDIs
and predicts protein-protein interactions based on protein
domains. The decision tree-based model is used to infer
domain–domain
interactions for each correctly predicted protein-protein interaction pair. The set of 2,475 DDIs
between Pfam-A domains was used. |
|
K-GIDDI |
386 |
K-GIDDI uses gene ontology information to
construct an initial DDI network using the top s% of DDIs inferred
from cross-species protein interaction networks, and then expands the DDI
network by predicting additional interactions using a graph theoretical approach based
on a parameter b. The latter procedure allows for prediction of interactions that can
otherwise not predictable by methods that rely solely on protein interaction data.
The set of 386 interactions predicted using s=10 and b=50 was used. |
|
Insite |
2,408 |
Insite uses a naive Bayes model to build
upon features in DPEA. Its novel formulation of evidence models for protein interactions and
DDIs helps address noise (false-positives) generated by
high-throughput assays. The set of 2,408 interactions with score ≥ 1 was used.
|
|
DomainGA |
459 |
DomainGA is a genetic
algorithm-type machine learning approach based on multi-parameter optimization.
It uses the available protein interaction data to compute a score for every domain pair,
which are then used to predict protein interactions. Yeast protein interaction dataset
was used to identify 867 putative DDIs between domains defined
based on information derived from the Interpro database. The set of 459 interactions
only between Pfam domains was used.
|
|
DIMA |
8,.012 |
DIMA contains DDI predictions based on phylogenetic profiling.
A set of 8,012 interactions reported in Pagel et al.'s J Mol Biol
(2004)
paper was used. |
DOMINE contains a total of 26,219 DDIs out of which 6,634
(gold-standard positives) are
inferred from PDB entries (the union of the sets of interactions from
iPfam and 3did), and
21,620 are predicted by at least one out of the 13 computational approaches.
The confidence levels of predictions by each computational approach was computed based on
how well an approach's predictions are also confirmed by other approaches. For every pair of
methods, Jaccard index values were computed.
Then, these were used to compute prediction overlap index(POI) for each method.
The figure below shows the schematic overview of the DOMINE database construction including
the new classification scheme to assign confidence levels to each DDI. The POI scores for 12 methods
are shown as a table within this figure. A method whose predictions do not overlap with
those of any other methods will receive a POI of one, whereas a method whose predictions overlap
completely with those of at least one other method will receive a POI of not more than 0.5. Thus,
a high POI for a method indicates this methods predictions are mostly either outliers
(false-positives) or novel putative DDIs. The confidence score for each predicted DDI is computed
by summing up the POIs of methods predicting. Based on a predicted DDI's confidence score and gene
ontology information on the interacting domains, it is either classified as either a high-confidence,
medium-confidence, or a low-confidence prediction (HCP, MCP, and LCP, respectively). We made a
decision to classify predictions by the ME approach HCPs since its predictions are based on
three sufficiently different types of information (protein interaction data, gene ontology
and gene fusion), and over 50% of them are known to be true.
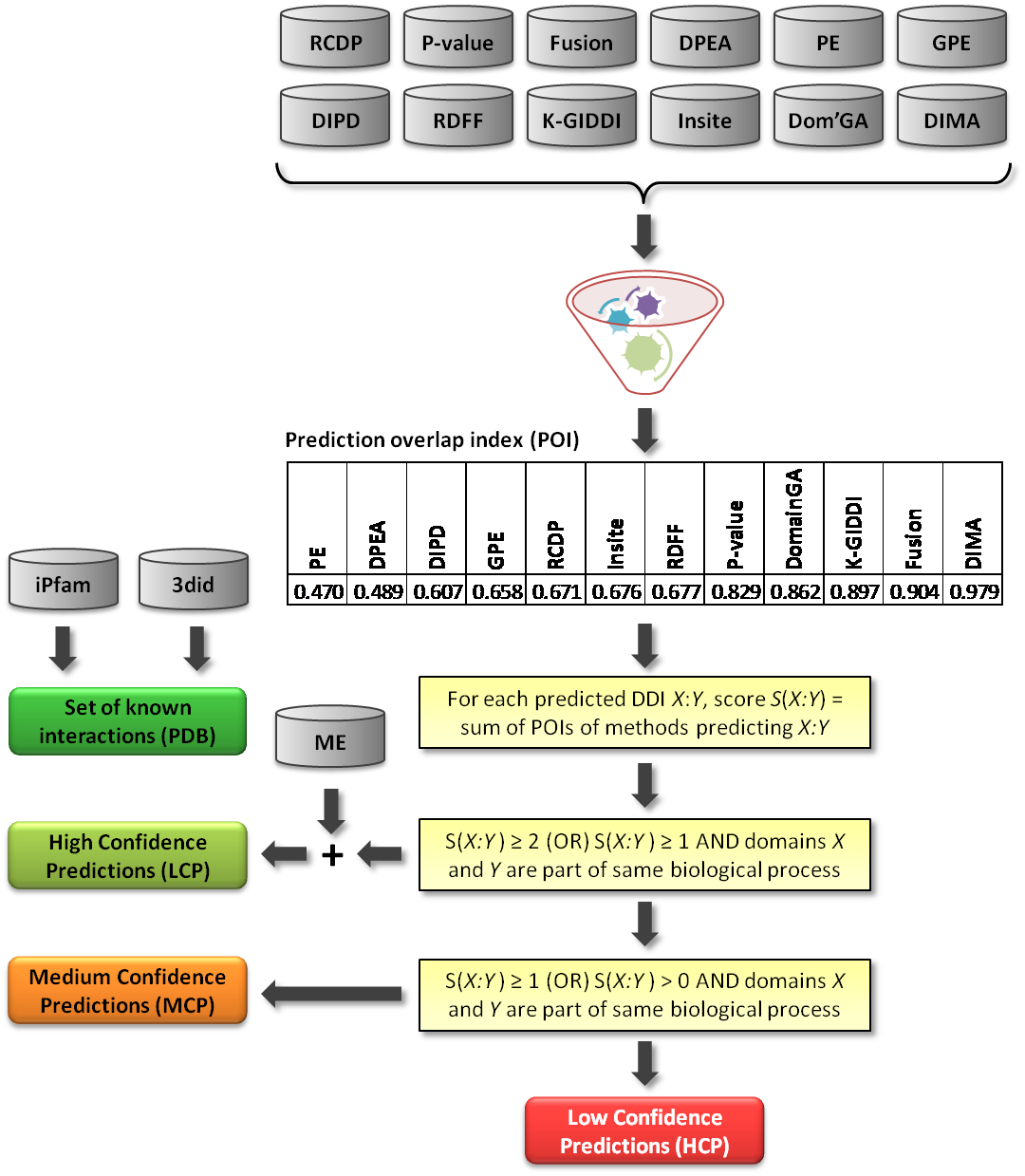
This new scheme resulted in 2,989, 2,537, and 16094 DDIs classified as HCP, MCP, and LCP, respectively.
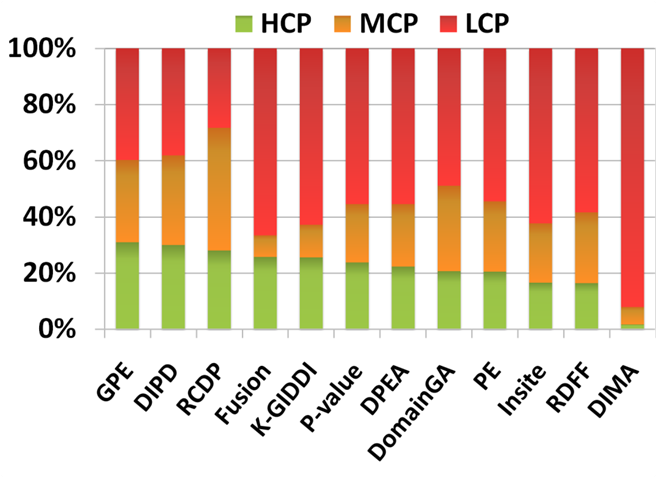
The following figure shows the percentages of predictions by each method classified as HCP, MCP, or LCP
B. Getting Started
How to use the database?
Domain-domain interaction information contained in the DOMINE database can
be accessed by clicking either the "Browse" or the "Search" option on the
menu.
Users who wish to browse the database have the option to browse a list of
Pfam domain IDs based on their gene ontology (GO) classification.
Users who wish to search the database may perform their search using
either a keyword (e.g., Insulin) or Pfam domain ID (e.g., Insulin) or
accession (e.g., PF00049 or 00049). Users may also search the database
using Interpro (e.g., IPR016179 or 016179 or 16179) and GO (e.g.,
GO:0005179 or 0005179 or 5179) identifiers. If you wish to search using
multiple identifiers, be sure to separate those using spaces. For
convenience, a search toolbar is provided right within the menu bar
(top-right corner of the Web Site).
What do the results mean?
Clicking on a domain name (Pfam ID) from anywhere on the Web Site displays
interaction information, if available, for that domain. For each
interacting domain, the list of domains that it is known/predicted to
interact with are displayed along with external links to the Pfam,
Interpro, and GO databases. For each predicted interaction, information on
whether DOMINE considers it to be a high-, medium-, or low-confidence
prediction is provided in addition to the source(s) of evidence.
Notes
 |
Interaction
observed in PDB crystal structure(s).
|
 |
Predicted interaction with either confidence score ≥ 2,
or confidence score ≥ 1 and the domains share a ontology term (part of the same biological process). |
 |
Predicted interaction with either confidence score ≥ 1, or the domains share
a gene ontology term (part of the same biological process). |
 |
Predicted interaction that is not classified as HCP or MCP. |
Source
 |
Interaction
observed in PDB crystal structure(s),
as inferred by
iPfam
|
 |
Interaction
observed in PDB crystal structure(s),
as inferred by
3did |
 |
Interaction predicted by
Lee et al.'s
integrated approach |
 |
Interaction predicted by
Jothi et al.'s using sequence co-evolution |
 |
Interaction predicted by
Nye et al.'s statistical approach |
 |
Interaction predicted using domain fusion
hypothesis, as inferred
by
Interdom |
 |
Interaction predicted by
Riley et al.'s using domain pair exclusion analysis |
 |
Interaction predicted by
Guimaraes et al.'s based on parsimonious explanation argument |
 |
Interaction predicted by
Guimaraes and Przytycka's generalized parsimonious explanation argument |
 |
Interaction predicted by
Zhao et al.'s discriminative classifier approach |
 |
Interaction predicted by
Chen and Liu's random forest algorithm |
 |
Interaction predicted by
Liu et al.'s graph-theoretical approach based on gene ontology data |
 |
Interaction predicted by
Wang et al.'s naive Bayes model |
 |
Interaction predicted by
Singhal and Resat's genetic algorithm-type machine learning approach |
 |
Interaction predicted by
Pagel et al.'s phylogenetic profiling approach |
C. Terms of Use
-
The following terms and conditions apply to all those who
use the DOMINE database or this Web Site. By visiting, accessing, browsing and/or using this
web site, you acknowledge that you have read, understood, and agree, to be
bound by these terms and conditions. If you do not agree to these terms,
do not use this web site.
-
It is forbidden to redistribute any of the Content of this Web Site
in any manner or create a database in electronic form or manually by
downloading and storing any such content without an express written
consent from the DOMINE team. Moreover, it is forbidden to sell any
information derived from this Web Site.
-
You may not make copies or mirrors of the DOMINE
database without a prior authorization from the DOMINE team.
-
The DOMINE team do not assume any legal liability or
responsibility for the quality, accuracy, truth, suitability,
completeness, or usefulness of any data, material or other information
contained on this Web Site.
-
The DOMINE team reserves the right to change these
terms and conditions by posting changes on this page of the Web Site and
you will be deemed to have accepted such changes if you use this Web Site
after the DOMINE team has published the amended terms and conditions on
this page of the Web Site.
D. Download Data
Click here to download data from
the DOMINE database.
E. Citing DOMINE
- Yellaboina S, Tasneem A, Zaykin DV, Raghavachari B, and Jothi R. DOMINE: A comprehensive collection of known and predicted domain-domain interactions, Nucleic Acids Research, Vol 39 (Database Issue), D730-735, 2011.
- Raghavachari B, Tasneem A, Przytycka, T, and Jothi R. DOMINE: A database of protein domain interactions, Nucleic Acids Research, Vol 36 (Database Issue), D656-661, 2008.
F. Frequently Asked Questions
1. What is DOMINE?
DOMINE is a database of protein domain (domain-domain) interactions
inferred from PDB entries, and those
that are predicted by
8 different computational approaches using
Pfam domain definitions.
2. What is a domain?
Domain is a structural or functional subunit of a protein. The concept of
the domain was first proposed in 1973 by Wetlaufer, who defined domains as
stable units of protein structure that could fold autonomously. We refer
the reader to
Wikipedia to learn more about domains.
3. Which domain definition is being use by DOMINE?
DOMINE uses domains as defined by the
Pfam.
4. What is the difference between a protein-protein interaction and a
domain-domain interaction?
Interaction between two proteins are referred to as a protein-protein
interaction. Domain is a structural or functional subunit of a protein,
and Interaction between two proteins often involves binding between pair(s)
of their constituent domains. Interaction between two domains of the same
protein are referred to as intra-chain domain-domain interaction, and
interaction between two domains from different proteins are referred to as
inter-chain domain-domain interaction.
5. DOMINE does not contain interaction information (or partners) for
YYYY domain!
None of the 10 data sources we used inferred YYYY domain to be interacting
with any other domain. It is possible that YYYY domain may actually be
interacting with other domains. If you know of such an interaction, and
have supporting information, please write to us. We would like to be
alerted on new domain-domain interactions, and would be happy to add them
to the database after examination.
6. Can I search my protein against DOMINE?
No. Unfortunately, the current version of DOMINE does not have
that capability. However, the domain composition of the protein
of interest can be obtained using
Pfam search,
and these domains can be searched against DOMINE.
|